

新闻应用
更新时间:2024-09-25 浏览次数:548
疫苗的生产属于无菌药品生产,疫苗生产企业在生产过程中要遵循2010年修订版GMP。这要求疫苗生产企业做到以下几个必须:
必须在合格供应商处采购原材料;
必须使用验证合格的厂房设施设备;
必须保证工作人员持续培训考核;
必须遵循药监部门批准的工艺生产;
必须做好各环节产成品的检定放行并按要求存储。
所有这些环节,还必须做到事无巨细真实完整的记录,这样才能保证生产出的每一支疫苗都是合格的产品。

疫苗生产的配制、过滤、转运、灌装等过程中,生产环境洁净级别须在B级背景下的A级(ISO 4 .8)操作。那么A级的洁净级别代表什么意思呢?A级,即不允许检出细菌,也就是表面微生物<1cfu/碟、沉降菌<1cfu/4小时、每立方米空气中的浮游菌<1cfu/m3!这是一个相当严苛的标准。
在2010版GMP附录1无菌药品第十一条中提到应当对微生物进行动态监测,评估无菌生产的微生物状况。监测方法包括沉降菌法、定量空气浮游菌采样法和表面取样法(如棉签擦拭法和接触碟法)等。动态取样应当避免对洁净区造成不良影响。成品批记录的审核应当包括环境监测的结果。

其中表面菌和沉降菌对采样设备要求不高,在这里不做讨论。定量空气浮游菌采样法之所以能够定量,是因为浮游菌采样器采样是一个主动过程,其可以收集一定体积空气中的微生物,能够更准确的反映洁净区域微生物的污染水平,因此在洁净室环境监测控制中得到广泛应用。

如何选择浮游菌采样器
根据GB/T 16293里面的描述,浮游菌采样器按采样原理分为狭缝式、离心式和针孔式三种,其中针孔式采样器是市场上常见的。
由于GB 16293对浮游菌采样器的描述也不详尽,我们主要参考ISO 14698,其描述为:
采样速率
采样装置至少要符合以下要求:
a)在适当时间内采集1m3、而又不会使采样培养基明显干燥的足够流量,如约100L/min;
b)对培养基的中等震动程度,如<20m/s
采样器特征
采样器的特性,如:
1)对于低含量空气悬浮活粒子的适当采吸流量
2)适当的震动/气流速度
3)采样精度与效率
4)易于搬运(重量,体积)和操作(使用方便,辅助设备,对真空泵的依赖,水、电等)
5)易于清洗、消毒或灭菌
6)对要测量的生物污染不会自然增加活粒子
来自采样器的排气不应污染洁净室

单向层流下等动力要求
有源细菌采样器

采样器采样效率评估


浮游菌如何采样
采样点的设置
采样点设置一般参考GB/T16293,具体如下:


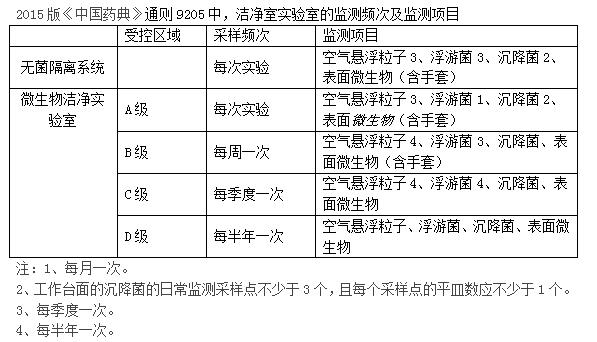
采样频次
药典通则9205中有对采样频次进行相关描叙:
采样量设定
单个位点单次小采样量参见GMP附录:

数据采集
相关的采样数据应当准确、可靠,要符合药企生产检验所遵循的“ALCOA+CCEA”原则。
一般实验人员习惯将采样数据以及培养计数数据记录在实验本上,若需要电子存档还要转誊至电脑,这中间不可控的人为因素较多,也是容易进行删改的地方,这种记录方式在要求更严格的欧洲其实已经不推荐了。

2018年3月9日,MHRA发布的《GxP数据完整性指南和定义》中就提到系统和流程的设计方式应有助于符合数据完整性原则。促成所需行为的内容包括但不限于:在活动发生的地点可以获得记录,这样不会有非正式的数据记录然后再转誊至正式记录的情况发生。防止未经授权的数据修改的用户访问权限(或审计跟踪,如果不能防止的话)。使用外部设备或系统接口方法,消除手工数据输入和与计算机系统的人工交互,如条形码扫描器、身份证阅读器或打印机。

另一机构APIC (欧洲化学工业委员会分支机构),发布了一系列针对原料药的GMP指南), 其9月份发布的《基于数据完整性管理实践指南》里就建议公司应从技术、程序和行为方面确保数据质量,尽可能自动捕获GXP数据,所以扫码自动获得采样数据或许是以后的发展趋势。
意大利ORUM浮游菌采样仪已有四十余年生产设计制造经验,其先后参与过和平号空间站环境监测项目和欧洲细菌战防御项目,产品质量值得信赖。其齐全的产品线,不管是单头/双头/三头采样器,还是隔离器专用或压缩气体专用采样器都可提供,完全满足ISO 14698的设计验证标准。

 微生物科研
微生物科研 微生物检测
微生物检测 微元素分析
微元素分析 解决方案
解决方案